



Содержание
Перейти к:
 Е. В. Кудрявцева,
О. В. Лагутина,
В. В. Ковалев,
С. С. Дерябина,
И. А. Захарова,
А. Ю. Черепенникова
Е. В. Кудрявцева,
О. В. Лагутина,
В. В. Ковалев,
С. С. Дерябина,
И. А. Захарова,
А. Ю. Черепенникова https://doi.org/10.17749/2313-7347/ob.gyn.rep.2023.441
Перейти к:
Введение. Частота бесплодного брака составляет 17–24 % и имеет тенденцию к увеличению. С каждым годом все больше бесплодных пар обращаются к вспомогательным репродуктивным технологиям (ВРТ). Если результат достигнут, будущие родители готовятся к появлению в семье здорового ребенка, но каждая пара может столкнуться с рождением потомства с тяжелым генетическим заболеванием. Одним из таких заболеваний является спинальная мышечная атрофия (СМА).
Цель: оценить частоту носительства делеции 7-го экзона в гене SMN1, ассоциированного со СМА, и количество копий гена SMN2 в супружеских парах, планирующих деторождение с помощью экстракорпорального оплодотворения (ЭКО).
Материалы и методы. В исследование вошли 170 супружеских пар (340 человек), страдающих бесплодием и направленных на проведение ЭКО в Свердловской области (СО) Российской Федерации. Проводился поиск делеций/дупликаций в генах SMN1 и SMN2 количественным анализом числа копий генов с использованием коммерческого набора SALSA MLPA Probemix P460 (MRC-Holland, Нидерланды). Для расчета предполагаемой частоты гомозиготных носителей делеций SMN1 в следующем поколении использовалось соотношение Харди–Вайнберга.
Результаты. Среди 340 пациентов делеция 7 экзона гена SMN1 (наличие одной копии из двух) обнаружена у 9 человек (3 мужчин и 6 женщин), не состоящих в супружеских отношениях между собой. Таким образом, всего 9 из 340 обследованных являются носителями мутации, ассоциированной с СМА, суммарная частота носительства составила 2,65 % (1/38). Учитывая количество проводимых в СО процедур ЭКО, можно предположить, что вероятность рождения больного ребенка в такой паре составляет не менее 1:6410. У ряда пациентов были выявлены дупликации в гене SMN1 – 9 (5,29 %) мужчин и 4 (2,35 %) женщины имели по 3 копии гена SMN1. Также было исследовано количества копий гена SMN2. Большинство участников исследования (54 %) имели 3 копии гена.
Заключение. Частота носительства СМА у супружеских пар, планирующих беременность с применением ВРТ, соответствует общепопуляционной и составляет 1:38 человек. Считаем необходимым, чтобы все супружеские пары, вступающие в программу ЭКО, были обследованы на носительство мутаций в гене SMN1 для определения риска наличия заболевания у потомства.
Кудрявцева Е.В., Лагутина О.В., Ковалев В.В., Дерябина С.С., Захарова И.А., Черепенникова А.Ю. Молекулярно-генетическое исследование ассоциированных со спинальной мышечной атрофией генов SMN1 и SMN2 у лиц с бесплодием, направленных на проведение экстракорпорального оплодотворения. Акушерство, Гинекология и Репродукция. 2023;17(6):707-717. https://doi.org/10.17749/2313-7347/ob.gyn.rep.2023.441
Kudryavtseva E.V., Lagutina O.V., Kovalev V.V., Deryabina S.S., Zakharova I.A., Cherepennikova A.Yu. Molecular-genetic study of SMN1 and SMN2 genes associated with spinal muscular atrophy in individuals with infertility prior to in vitro fertilization. Obstetrics, Gynecology and Reproduction. 2023;17(6):707-717. (In Russ.) https://doi.org/10.17749/2313-7347/ob.gyn.rep.2023.441
Бесплодный брак является одной из самых актуальных проблем современного общества. В настоящее время в клиниках репродуктивных технологий с диагнозом «бесплодие» наблюдаются примерно 186 млн человек во всем мире [1]. С течением времени ситуация только усугубляется. Распространенность данного заболевания продолжает расти, особенно в развитых странах. В России, по оценкам разных экспертов, частота бесплодия составляет от 17 до 24 % супружеских пар [2]. На фоне этого все более востребованными становятся вспомогательные репродуктивные технологии (ВРТ) как последняя надежда на появление желанного ребенка в семье. В России, по данным Регистра ВРТ Российской ассоциации репродукции человека (РАРЧ), в 2020 г. было выполнено 148 660 различных процедур ВРТ, в результате чего родилось около 31000 детей. Ежегодно число процедур ВРТ продолжает увеличиваться [3].
Средняя результативность экстракорпорального оплодотворения (ЭКО) составляет около 30 % (при различных причинах бесплодия) [3]. Обычно требуется более одной попытки ВРТ. При положительном результате – наступлении долгожданной беременности – супруги готовятся к появлению на свет здорового ребенка, но каждая пара может столкнуться с рождением потомства с генетическим заболеванием. Одним из таких заболеваний является спинальная мышечная атрофия (СМА) – генетически гетерогенная группа заболеваний, характеризующихся прогрессирующей дегенерацией и гибелью двигательных нейронов передних рогов спинного мозга, а также в ряде случаев ядер ствола головного мозга [4–6]. Данное заболевание рассматривается как одна из ведущих причин смертности от наследственной патологии у детей с дебютом в раннем возрасте [7]. Повышенное внимание к проблемам СМА также обусловлено неуклонным увеличением числа таких пациентов в последние десятилетия [8][9].
Общемировые данные по частоте носительства данного заболевания колеблются на цифрах 1/40–1/50 [10–13], при этом наиболее высокая частота носительства отмечена у европеоидов, а наименьшая – у афроамериканцев [14]. По данным ФГБНУ МГНЦ им. акад. Н.П. Бочкова, общероссийская частота носительства патогенного варианта в гене SMN1 составляет 1/36 человек [11]. Исходя из этого, количество носителей данной наследственной патологии среди пациентов, планирующих беременность с помощью ВРТ, можно ожидать на уровне 3–5 тыс. человек ежегодно.
В настоящее время существует возможность проведения предимплантационного генетического тестирования (ПГТ) в программах ЭКО перед переносом эмбриона в полость матки. Такое исследование включает в себя целый комплекс методов для получения генетического материала будущего плода и диагностики у него анеуплоидий (ПГТ-А), структурных хромосомных перестроек (ПГТ-СП) и моногенных мутаций (ПГТ-М) [3][8]. Главное преимущество тестирования эмбрионов на доимплантационном этапе заключается в том, что это значительно снижает риск обнаружения генетической патологии у плода при уже наступившей беременности, тем более если она была осуществлена с помощью ВРТ. В настоящее время специалисты все чаще предлагают рассматривать ПГТ как обязательную составляющую превентивной медицины, поскольку это самая ранняя форма пренатального тестирования [15].
К сожалению, на практике к данному методу прибегают не часто, а если проводят диагностику, то в основном применяют ПГТ-А. ПГТ-М проводится только в тех случаях, когда достоверно установлено носительство определенного патогенного генетического варианта у обоих супругов. Статус же носительства рецессивных заболеваний устанавливается в большинстве случаев лишь после рождения в семье больного ребенка. Решением данной проблемы вполне может стать преконцепционный скрининг – тестирование будущих родителей на гетерозиготное носительство моногенных заболеваний. Так, Американский колледж медицинской генетики и геномики (англ. American College of Medical Genetics and Genomics, ACMG) рекомендует популяционный скрининг на носительство делеций в гене SMN1 у лиц репродуктивного возраста из-за высокой частоты носительства и непомерной стоимости терапии [16]. Американская ассоциация акушеров и гинекологов (англ. American College of Obstetricians and Gynecologists, ACOG) также рекомендует проводить скрининг на носительство СМА всем беременным и супружеским парам, независимо от их национальности [17]. При этом семейные пары, планирующие беременность с помощью процедуры ЭКО, как правило, остаются без внимания. Учитывая рост количества детей, ежегодно появляющихся на свет в результате ВРТ, и частоту распространенности СМА, проблема предупреждения рождения детей с данной патологией в бесплодном браке является актуальной и заслуживает как научного, так и практического интереса.
Цель: оценить частоту носительства делеции 7-го экзона в гене SMN1, ассоциированного со СМА, и количество копий гена SMN2 в супружеских парах, планирующих деторождение с помощью ЭКО.
В исследование вошли 170 супружеских пар (340 человек), страдающих бесплодием и направленных на подготовку к родам по программе ЭКО. Все участники исследования обследовались в ГАУЗ СО «Клинико-диагностический центр "Охрана здоровья матери и ребенка"», все были жителями Свердловской области Российской Федерации (РФ) и не имели в родословных сведений о моногенных заболеваниях.
Критерии включения: пациенты, направленные на ЭКО в связи с бесплодием по программе обязательного медицинского страхования, имеющие заключение о нормальном кариотипе у обоих супругов.
Критерии невключения: наличие показаний для использования донорских гамет в программах ВРТ; аномальный кариотип у одного или обоих супругов.
Критерии исключения: отказ супругов от участия в исследовании.
Исследование проведено в соответствии с этическими стандартами Хельсинской декларации Всемирной медицинской ассоциации 1964 г. и ее последующими изменениями. Все пациенты подписали добровольное информированное согласие на участие в исследовании и отбор биоматериала для анализа. Исследование одобрено на заседании этического комитета ГБУЗ СО «Екатеринбургский клинический перинатальный центр» (протокол № 2 от 26.05.2023).
Всем пациентам было проведено до- и послетестовое медико-генетическое консультирование. На дотестовом консультировании пациентам объясняли преимущества и ограничения проводимого исследования, пациенты подписывали информированное согласие, после чего проводили отбор биологического материала. При послетестовом консультировании разъяснялись результаты исследования.
Геномную ДНК из образцов цельной крови выделяли автоматическим методом на станции MagNa Pure LC 2.0 (Roche, США). Поиск делеций/дупликаций в генах SMN1 и SMN2 проводили количественным анализом числа копий генов с использованием коммерческого набора SALSA MLPA Probemix P460 (MRC-Holland, Нидерланды). Анализ образцов проводили согласно инструкции фирмы-производителя на генетическом анализаторе Applied Biosystems 3500 (Thermo Fisher Scientific, США) с использованием аналитического программного обеспечения Coffalyser.net (MRC-Holland, Нидерланды).
Статистическая обработка проводилась с помощью программы StatPlus 8.0.3. (2021, AnalystSoft Inc.). Для количественных показателей указывалась медиана с интерквартильным размахом (Ме [Q1; Q3]). Качественные показатели указывались в абсолютных и относительных величинах. Для расчета предполагаемой частоты гомозиготных носителей в следующем поколении использовалось соотношение Харди-Вайнберга.
Клинико-анамнестическая характеристика обследованных / Clinical and anamnestic characteristics of patients examined
Средний медианный возраст обследованных женщин составил 34 [ 31; 37] года, мужчин – 36 [ 33; 38] лет. Продолжительность бесплодия в браке в среднем составляла 5 [ 2; 8] лет. Структура причин бесплодия представлена на рисунке 1.
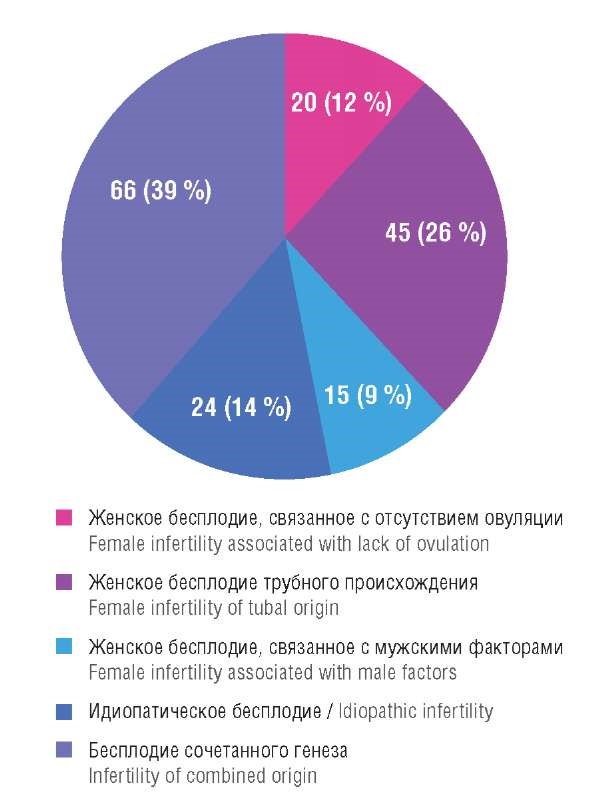
Рисунок 1. Структура причин бесплодия у обследованных супружеских пар.
Figure 1. Pattern of infertility-related causes in the examined couples.
Из всех обследованных 63 пары (37 %) обратились за помощью к ВРТ впервые, 58 пар (34,1 %) имели одну попытку ЭКО, либо ЭКО+ИКСИ (интрацитоплазматическая инъекция сперматозоида) в анамнезе, 33 пары (19,5 %) имели 2 таких попытки, остальные 16 (9,5%) участвовали в программе ЭКО более 3 раз; в 50 (29,4 %) супружеских парах на момент обследования имелись здоровые дети.
Графическое изображение результата тестирования показано на рисунке 2. Общая схема распределения копийности генов SMN в нашей выборке представлена в таблице 1.

Рисунок 2. Геномные координаты фрагментов ДНК, исследованных
с помощью набора SALSA MLPA Probemix P460-A1 SMA (Silent) Carrier.
Примечание: А – две копии гена SMN1;
В – три копии гена SMN1;
С – одна копия гена SMN1.
Figure 2. Genomic coordinates of DNA fragments examined
with the SALSA MLPA Probemix P460-A1 SMA (Silent) Carrier kit.
Note: A – two copies of SMN1 gene;
В – three copies of SMN1 gene;
С – one copy of SMN1 gene.
Таблица 1. Распределение генотипов 340 пациентов по количеству копий генов SMN.
Table 1. Distribution of genotypes in 340 patients related to SMN gene copies number.
|
Ген / Gene |
Количество копий / Сopy number |
Всего / Total n = 340 n (%) |
Мужчины / Male n = 170 n (%) |
Женщины / Female n = 170 n (%) |
|
SMN1 |
1 |
9 (2,64) |
3 (1,76) |
6 (3,53) |
|
2 |
318 (93,53) |
158 (92,94) |
160 (94,12) |
|
|
3 |
13 (3,82) |
9 (5,29) |
4 (2,35) |
|
|
SMN2 |
0 |
26 (7,65) |
13 (7,65) |
13 (7,65) |
|
1 |
124 (36,47) |
57 (33,53) |
67 (39,41) |
|
|
2 |
182 (53,53) |
98 (57,65) |
84 (49,41) |
|
|
3 |
8 (2,35) |
2 (1,18) |
6 (3,53) |
Определяющим фактором при тестировании на гетерозиготное носительство служит установление количества копий 7-го экзона гена SMN1. Среди 340 пациентов делеция данного участка ДНК (наличие одной копии из двух) обнаружена у 9 человек (3 мужчин и 6 женщин), не состоящих в супружеских отношениях между собой. Таким образом, всего 9 из 340 обследованных являются носителями мутации, ассоциированной с СМА, суммарная частота носительства составила 2,65 % (1/38).
Расчёт частоты мутантного (q) и нормального аллея (р) гена SMN1 среди мужчин (m) и женщин (f) по теории равновесия Харди-Вайнберга [18] дает нам представление о частоте гомозиготного носительства делеции (q2) в популяции, на основании которого мы можем предполагать, что частота заболевания СМА в следующем поколении составит 1,56 на 10 тыс. (1:6410). Результаты расчетов представлены в таблице 2.
Таблица 2. Расчет частоты различных генотипов SMN1 в следующем поколении.
Table 2. Calculated rate for various next generation SMN1 genotypes.
|
f/m |
р = 0,991177 |
q = 0,008823 |
|
p = 0,982352 |
p² = 0,973685 |
pq = 0,008667 |
|
q = 0,017647 |
pq = 0,017491 |
q² = 0,000156 |
У ряда пациентов были выявлены дупликации в гене SMN1 – 9 (5,29 %) мужчин и 4 (2,35 %) женщины имели по 3 копии гена SMN1.
Также было исследовано количества копий гена SMN2. Результаты представлены на рисунке 3. Большинство участников исследования (54 %) имели 3 копии гена.
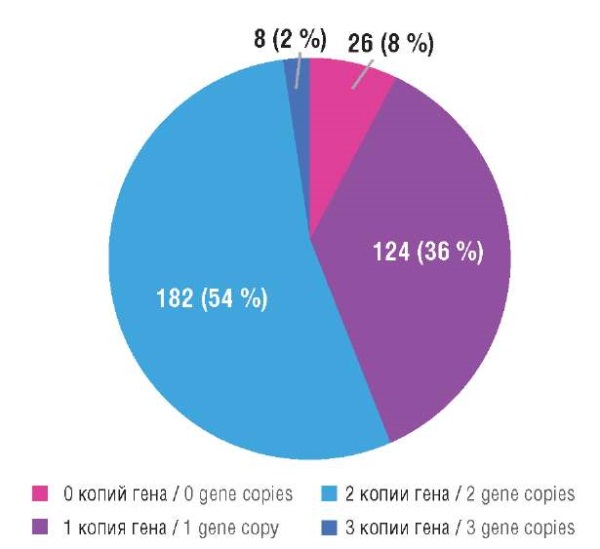
Рисунок 3. Число копий гена SMN2 у участников исследования.
Figure 3. Number of SMN2 gene copies in study participants.
В нашем исследовании впервые был проведен анализ частоты носительства делеции 5q SMN1 именно среди пациентов, направленных на ЭКО, которым может быть предложено предимплантационное генетическое тестирование эмбрионов.
По нашим расчетам, суммарная частота носительства среди участников исследования составила 1/38 (2,65 %). Аналогичные данные в РФ были получены ранее [11]. В целом, при оценке распространенности носительства делеций в гене SMN1 в мире эта цифра немного ниже – 1/40–1/50 (2,0–2,5 %) [10][12][13].
В соответствии с нашими расчетами, частота СМА в следующием поколении детей, рожденных с помощью ВРТ, составит 1,5:10000.
Исходя из открытых данных РАРЧ [3], в РФ каждый год появляется на свет около 30 тыс. детей, рожденных в результате применения ЭКО, и это значит, что потенциально 4 ребенка из них будут иметь диагноз СМА. Напомним, что данное заболевание является одной из наиболее распространенных генетических причин младенческой смертности [12][19][20], и несмотря на существующее в настоящее время патогенетическое лечение данного заболевания, вопрос о полном излечении таких пациентов, особенно имеющих СМА тип 0, пока не стоит [21].
В РФ и за рубежом были проведены исследования социально-экономической нагрузки СМА. В каждом из них авторы делают выводы, что экономические расходы на лечение больных СМА детей огромны, а следовательно, стоит рассмотреть вопрос о введении скрининга на носительство мутаций, ассоциированных с СМА, супружеским парам, планирующим деторождение [20][22][23]. Результаты российских исследований также показали, что СМА в РФ сопряжена с существенными материальными затратами. По данным А.С. Колбина с соавт. на 2020 г., затраты составили 5,44 млрд руб/год. Анализ показывает, что по мере расширения доступа пациентов к патогенетической терапии, бремя СМА будет возрастать [24]. При этом экономическую эффективность преконцепционного скрининга носительства наследственных заболеваний, в том числе СМА, оценить достаточно сложно, так как расходы вычисляются не только финансовыми потерями на препараты для лечения, но и множеством других социальных и экономических нюансов.
В случае направления пациентов на ВРТ этот вопрос также крайне актуален, поскольку в рамках национального проекта «Демография» ежегодно направляются средства на проведение парам с бесплодием ЭКО за счет средств базовой программы обязательного медицинского страхования (ОМС). В 2024 г. в процессе реализации нацпроекта «Демография» планируется провести не менее 450 тыс. процедур ЭКО [25]. Если хотя бы каждая пятая из них закончится рождением ребенка, на счет появится около 90 тыс. детей. Учитывая частоту носительства делеций в гене SMN1, при отсутствии определения статуса носительства у родителей, 13–14 среди этих детей будут больны СМА. Выявление патогенных генетических вариантов у супругов на этапе преконцепционной подготовки дает возможность проведения предимплантационного генетического тестирования эмбрионов (ПГТ-М).
Важной информацией является определение числа копий SMN2, поскольку это может существенно влиять на тяжесть симптомов у больного, хотя сами по себе изменения в гене SMN2 не могут быть причиной СМА и не имеют клинического значения, если не нарушена функция белка SMN1.
Согласно научным данным, количество копий гена SMN2 может варьировать от 0 до 8 [26]. Количество копий гена SMN2 у здоровых лиц не имеет клинического значения, однако при наличии СМА тяжесть клинических проявлений коррелирует с этим параметром [26–28]. От количества копий гена SMN2 зависит и терапевтическая стратегия, так как он является основной мишенью для ряда препаратов (в том числе нусинерсен, рисдиплан) [7][10][26].
Кроме носителей делеции гена SMN1, в нашем исследовании обнаружены 12 (3,53 %) человек, имеющих дупликацию аналогичного участка генома. По мнению ряда авторов, подобные дупликации гена SMN1 могут быть связаны с предрасположенностью к амиотрофическому латеральному склерозу, и данная информация может быть использована при медико-генетическом консультировании [29–31]. Действительно, пациенты с тремя копиями SMN1 имеют более ранний средний возраст начала заболевания и большую продолжительность заболевания по сравнению с пациентами с двумя копиями SMN1 [30]. В то же время патогенность данного варианта не доказана, а учитывая частоту встречаемости данного варианта среди обследованных нами пар (3,52 %), этот вариант не является патогенным [32]. Поэтому наше мнение сводится к тому, что указывать данную особенность генотипа при выдаче заключения нецелесообразно [32].
Важным дополнением, на наш взгляд, можно считать отсутствие в представленной выборке генотипа «2+0» (носителей с двумя копиями гена SMN1 на одной хромосоме и без копий на второй хромосоме). Между тем такой вариант встречается в популяции с частотой примерно 0,087 % и может вызвать определенные трудности при диагностике СМА [33].
Несмотря на то что преконцепционный скрининг на носительство делеций 5q SMN1 помогает выявить группу высокого риска по рождению потомства, имеющего СМА, отрицательный результат тестирования не дает гарантии рождения ребенка без этой патологии. Как было сказано выше, в научной литературе описано носительство делеции 5q SMN1 у лиц, имеющих 2 копии гена SMN1 на одной хромосоме [33]. Кроме этого, делеции 5q SMN1 могут возникать de novo, что может приводить к наличию СМА у ребенка, у которого только один родитель является носителем аналогичной делеции [14]. Помимо делеции 5q, описаны точковые мутации гена SMN1, которые при гомозиготном или компаунд-гетерозиготном генотипе также могут приводить к развитию клинического фенотипа СМА различной степени тяжести [34–40]. В парах, где хотя бы у одного партнера выявлена одна копия SMN1, относительный риск рождения потомства с СМА выше популяционного, в то же время абсолютный риск остается низким и составляет менее 1 % [14]. И даже в случаях, когда делеция 5q не выявлена ни у одного из будущих родителей, риск не является нулевым [14].
В настоящее время в России отсутствуют какие-либо нормативные документы, касающиеся проведения преконцепционного генетического скрининга для определения риска для потомства СМА и других генетических заболеваний. Однако очевидно, что при внедрении подобных исследований в государственные программы, связанные с охраной здоровья населения, требуется обеспечение равноправного доступа к тестированию и соответствующим консультациям, а также обеспечение конфиденциальности и защиты частной информации [41][42]. При направлении на тестирование на носительство наследственных заболеваний перед применением ВРТ, при претестовом консультировании пациентам должно быть разъяснено, что даже в случае выявления патогенных генетических вариантов они не будут подвергнуты дискриминации при лечении бесплодия.
Ограничением нашего исследования было то, что мы проводили исследование только делеций 5q SMN1, но не исследовали точковые мутации в этом гене. В России частота носительства подобных генетических вариантов составляет 1:4464 [43]. Кроме того, наше исследование было проведено среди жителей Свердловской области, и мы не исключаем, что в других регионах РФ частота носительства делеций SMN1 может отличаться.
Частота носительства тяжелейшего генетического заболевания – спинальной мышечной атрофии – в категории супружеских пар, планирующих беременность с применением ВРТ, соответствует общепопуляционной и составляет 1:38 человек. Учитывая количество проводимых в Свердловской области процедур ЭКО, можно предположить, что вероятность рождения больного ребенка в такой паре составляет не менее 1:6410.
Исходя из этого, считаем необходимым, чтобы все супружеские пары, вступающие в программу ЭКО, были обследованы на носительство СМА для определения риска наличия заболевания у потомства. Впрочем, это в полной мере относится и ко всем другим категориям граждан, осознанно подходящих к своему будущему родительству.
1. Inhorn M.C., Patrizio P. Infertility around the globe: new thinking on gender, reproductive technologies and global movements in the 21st century. Hum Reprod Update. 2015;21(4):411–26. https://doi.org/10.1093/humupd/dmv016.
2. Клинические рекомендации – Женское бесплодие – 2021-2022-2023 (24.06.2021). М.: Министерство здравоохранения Российской Федерации, 2021. 50 с. Режим доступа: https://moniiag.ru/wp-content/uploads/2019/07/Klinicheskie-rekomendatsii.-ZHenskoe-besplodie.pdf. [Дата обращения: 11.06.2023].
3. Регистр ВРТ. Отчет за 2020 год. Российская ассоциация репродукции человека, 2020. 56 c. Режим доступа: https://rahr.ru/d_registr_otchet/RegistrVRT_2020.pdf. [Дата обращения: 11.06.2023].
4. Забненкова В.В., Дадали Е.Л., Поляков А.В. Проксимальная спинальная мышечная атрофия типов I–IV: особенности молекулярно генетической диагностики. Нервно-мышечные болезни. 2013;(3):27–31. https://doi.org/10.17650/2222-8721-2013-0-3-27-31.
5. Lunn M.R., Wang C.H. Spinal muscular atrophy. Lancet. 2008;371(9630):2120–33. https://doi.org/10.1016/S01406736(08)60921-6.
6. Prior T.W., Leach M.E., Finanger E. Spinal muscular atrophy. GeneReviews®. National Library of Medicine, 2020. Режим доступа: http://www.ncbi.nlm.nih.gov/books/NBK1352/. [Дата обращения: 11.06.2023].
7. Маретина М.А., Киселев А.В., Ильина А.В. и др. Современные тенденции в диагностике, скрининге и лечении спинальной мышечной атрофии. Вестник Российской академии медицинских наук. 2022;77(2):87–96. https://doi.org/10.15690/vramn1768.
8. Scriven P.N. Combining PGT-A with PGT-M risks trying to do too much. J Assist Reprod Genet. 2022;39(9):2015–8. https://doi.org/10.1007/s10815-022-02519-8.
9. Vill K., Blaschek A., Schara U. et al. Spinal muscular atrophy: Time for newborn screening? Nervenarzt. 2017;88(120:1358–66. (In German). https://doi.org/10.1007/s00115-017-0447-3.
10. Гузева В.И., Иванов Д.О., Петренко Ю.В. и др. Проксимальная спинальная мышечная атрофия 5q. Методическое пособие для врачей. СПб.: СПбГПМУ, 2021. 20 с.
11. Zabnenkova V.V., Dadali E.L., Spiridonova M.G. et al. Spinal muscular atrophy carrier frequency in Russian Federation. In: Proceedings of American Society of Human Genetics (ASHG). Annual Meeting, 2016. 2476W. https://doi.org/10.13140/RG.2.2.16245.60642.
12. Sugarman E.A., Nagan N., Zhu H. et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72,400 specimens. Eur J Hum Genet. 2012;20(1):27–32. https://doi.org/10.1038/ejhg.2011.134.
13. Ceylan A.C., Erdem H.B., Şahin İ., Agarwal M. SMN1 gene copy number analysis for spinal muscular atrophy (SMA) in a Turkish cohort by CODESEQ technology, an integrated solution for detection of SMN1 and SMN2 copy numbers and the “2+0” genotype. Neurol Sci. 2020;41:2575–84. https://doi.org/10.1007/s10072-020-04365-x.
14. Carré A., Empey C. Review of spinal muscular atrophy (SMA) for prenatal and pediatric genetic counselors. J Genet Couns. 2016;25(1):32–43. https://doi.org/10.1007/s10897-015-9859-z.
15. Плаксина А.Н., Ковтун О.П., Николаева Е.Б. Вспомогательные репродуктивные технологии: анализ достигнутых результатов и поиск новых решений (обзор литературы). Уральский медицинский журнал. 2017;(5):20–6.
16. Gates A., Terry S.F., Bonhomme N. Expanded carrier screening and its implications on genetic testing protocols. Genet Test Mol Biomarkers. 2016;20(11):643–4. https://doi.org/10.1089/gtmb.2016.29023.sjt.
17. Committee Opinion No. 690: Carrier Screening in the Age of Genomic Medicine. Obstet Gynecol. 2017;129(3):e35–40. https://doi.org/10.1097/AOG.0000000000001951.
18. Волобуев А.Н., Давыдкин И.Л., Колсанов А.В., Кудлай Д.А. Математические аспекты генетики. M.: ГЭОТАР-Медиа, 2020. 176 c. https://doi.org/10.33029/9704-5890-7-MAG-2020-1-176.
19. Hendrickson B.C., Donohoe C., Akmaev V.R. et al. Differences in SMN1 allele frequencies among ethnic groups within North America. J Med Genet. 2009;46(9):641–4. https://doi.org/10.1136/jmg.2009.066969.
20. Gillingwater T.H. Counting the cost of spinal muscular atrophy. J Med Econ. 2016;19(8):827–8. https://doi.org/10.1080/13696998.2016.1202833.
21. Невмержицкая К.С., Сапего Е.Ю., Морозова Д.А. Краткосрочная безопасность и эффективность онасемноген абепарвовека у 10 пациентов со спинальной мышечной атрофией: когортное исследование. Вопросы современной педиатрии. 2021;20(6s):589–94. https://doi.org/10.15690/vsp.v20i6S.2367.
22. Droege M., Sproule D., Arjunji R. et al. Economic burden of spinal muscular atrophy in the United States: a contemporary assessment. J Med Econ. 2020;23(1):70–9. https://doi.org/10.1080/13696998.2019.1646263.
23. Armstrong E.P., Malone D.C., Yeh W.-S. et al. The economic burden of spinal muscular atrophy. J Med Econ. 2016;19(8):822–6. https://doi.org/10.1080/13696998.2016.1198355.
24. Колбин А.С., Влодавец Д.В., Курылев А.А. и др. Анализ социально-экономического бремени спинальной мышечной атрофии в Российской Федерации. ФАРМАКОЭКОНОМИКА. Современная фармакоэкономика и фармакоэпидемиология. 2020;13(4):337–54. https://doi.org/10.17749/2070-4909/farmakoekonomika.2020.068.
25. Национальный проект «Демография». Режим доступа: https://mintrud.gov.ru/ministry/programms/demography. [Дата обращения: 11.06.2023].
26. Butchbach M.E.R. Genomic variability in the survival motor neuron genes (SMN1 and SMN2): Implications for spinal muscular atrophy phenotype and therapeutics development. Int J Mol Sci. 2021;22(15):7896. https://doi.org/10.3390/ijms22157896.
27. Клинические рекомендации. Проксимальная спинальная мышечная атрофия 5q. М.: Министерство здравоохранения Российской Федерации, 2023. 117 c. Режим доступа: https://amg-genetics.ru/pdf/2023/kr_sma_2023.pdf. [Дата обращения: 11.06.2023].
28. Rouzier C., Chaussenot A., Paquis-Flucklinger V. Molecular diagnosis and genetic counseling for spinal muscular atrophy (SMA). Arch Pediatr. 2020;27(7S):9–14. https://doi.org/10.1016/S0929-693X(20)30270-0.
29. Blauw H.M., Barnes C.P., van Vught P.W.J. et al. SMN1 gene duplications are associated with sporadic ALS. Neurology. 2012;78(11):776–80. https://doi.org/10.1212/WNL.0b013e318249f697.
30. Kuźma-Kozakiewicz M., Jędrzejowska M., Kaźmierczak B. SMN1 gene duplications are more frequent in patients with progressive muscular atrophy. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14(5–6):457–62. https://doi.org/10.3109/21678421.2013.771367.
31. Wang X.-.B, Cui N.-H., Gao J.-J. et al. SMN1 duplications contribute to sporadic amyotrophic lateral sclerosis susceptibility: evidence from a meta-analysis. J Neurol Sci. 2014;340(1–2):63–8. https://doi.org/10.1016/j.jns.2014.02.026.
32. Рыжкова О.П., Кардымон О.Л., Прохорчук Е.Б. и др. Руководство по интерпретации данных последовательности ДНК человека, полученных методами массового параллельного секвенирования (MPS) (редакция 2018, версия 2). Медицинская генетика. 2019;18(2):3–23. https://doi.org/10.25557/2073-7998.2019.02.3-23.
33. Ar Rochmah M., Awano H., Awaya T. et al. Spinal muscular atrophy carriers with two SMN1 copies. Brain Dev. 2017;39(10):851–60. https://doi.org/10.1016/j.braindev.2017.06.002.
34. Забненкова В.В., Дадали Е.Л., Артемьева С.Б. и др. Точковые мутации в гене SMN1 у больных проксимальной спинальной мышечной атрофией I–IV типа, имеющих одну копию гена SMN1. Генетика. 2015;51(9):1075–82. https://doi.org/10.7868/S0016675815080123.
35. Theodorou L., Nicolaou P., Koutsou P. et al. Genetic findings of Cypriot spinal muscular atrophy patients. Neurol Sci. 2015;36(10):1829–34. https://doi.org/10.1007/s10072-015-2263-5.
36. Souček .P, Réblová K., Kramárek M. et al. High-throughput analysis revealed mutations’ diverging effects on SMN1 exon 7 splicing. RNA Biol. 2019;16(10):1364–76. https://doi.org/10.1080/15476286.2019.1630796.
37. Sneha P., Zenith T.U., Abu Habib U.S. et al. Impact of missense mutations in survival motor neuron protein (SMN1) leading to Spinal Muscular Atrophy (SMA): A computational approach. Metab Brain Dis. 2018;33(6):1823–34. https://doi.org/10.1007/s11011-018-0285-4.
38. Ganji H., Nouri N., Salehi M. et al. Detection of intragenic SMN1 mutations in spinal muscular atrophy patients with a single copy of SMN1. J Child Neurol. 2015;30(5):558–62. https://doi.org/10.1177/0883073814521297.
39. Zhao X., Wang Y., Mei S. et al. Identification of two novel SMN1 point mutations associated with a very severe SMA-I phenotype. Eur J Med Genet. 2020;63(9):104006. https://doi.org/10.1016/j.ejmg.2020.104006.
40. Wijaya Y.O.S., Ar Rohmah M., Niba E.T.E. et al. Phenotypes of SMA patients retaining SMN1 with intragenic mutation. Brain Dev. 2021;43(7):745–58. https://doi.org/10.1016/j.braindev.2021.03.006.
41. Кудрявцева Е.В. Философские, медицинские и юридические аспекты репродуктивной генетики. Уральский медицинский журнал. 2018;(13):54–7. https://doi.org/10.25694/URMJ.2018.13.46.
42. Ижевская В.Л., Баранова Е.Е. Информированное согласие при генетическом тестировании и скрининге. Медицинская генетика. 2022;21(4):16–24. https://doi.org/10.25557/2073-7998.2022.04.16-24.
43. Михальчук К.А., Забненкова В.В., Щагина О.А., Поляков А.В. Спектр минорных вариантов локуса SMN. Медицинская генетика. 2022;21(10):19–22. https://doi.org/10.25557/2073-7998.2022.10.19-22.
Кудрявцева Елена Владимировна – д.м.н., зав. Центральной научно-исследовательской лабораторией ФГБОУ ВО «Уральский ГМУ» МЗРФ; научный сотрудник лаборатории молекулярно-генетических исследований ГАУЗ СО «Институт медицинских клеточных технологий», Екатеринбург, Россия; врач-генетик отделения медико-генетического консультирования ГАУЗ СО «Клинико-диагностический центр "Охрана здоровья матери и ребенка"».
620028 Екатеринбург, ул. Репина, д. 3; 620026 Екатеринбург, ул. Карла Маркса, д. 22А; 620067 Екатеринбург, ул. Флотская, д. 52
Лагутина Ольга Викторовна – научный сотрудник лаборатории молекулярно-генетических исследований.
620026 Екатеринбург, ул. Карла Маркса, д. 22А; 620067 Екатеринбург, ул. Флотская, д. 52
Ковалёв Владислав Викторович – д.м.н., профессор, зав. кафедрой акушерства и гинекологии, трансфузиологии.
620028 Екатеринбург, ул. Репина, д. 3
Дерябина Светлана Степановна – к.б.н., ассистент кафедры акушерства и гинекологии, трасфузиологии ФГБОУ ВО «Уральский ГМУ» МЗРФ; научный сотрудник ГАУЗ СО «Институт медицинских клеточных технологий»; зав. лабораторией молекулярной диагностики ГАУЗ СО «Клинико-диагностический центр "Охрана здоровья матери и ребенка"».
620028 Екатеринбург, ул. Репина, д. 3; 620026 Екатеринбург, ул. Карла Маркса, д. 22А; 620067 Екатеринбург, ул. Флотская, д. 52
Захарова Илона Александровна – ординатор.
620028 Екатеринбург, ул. Репина, д. 3
Черепенникова Алёна Юрьевна – ординатор.
620028 Екатеринбург, ул. Репина, д. 3
Кудрявцева Е.В., Лагутина О.В., Ковалев В.В., Дерябина С.С., Захарова И.А., Черепенникова А.Ю. Молекулярно-генетическое исследование ассоциированных со спинальной мышечной атрофией генов SMN1 и SMN2 у лиц с бесплодием, направленных на проведение экстракорпорального оплодотворения. Акушерство, Гинекология и Репродукция. 2023;17(6):707-717. https://doi.org/10.17749/2313-7347/ob.gyn.rep.2023.441
Kudryavtseva E.V., Lagutina O.V., Kovalev V.V., Deryabina S.S., Zakharova I.A., Cherepennikova A.Yu. Molecular-genetic study of SMN1 and SMN2 genes associated with spinal muscular atrophy in individuals with infertility prior to in vitro fertilization. Obstetrics, Gynecology and Reproduction. 2023;17(6):707-717. (In Russ.) https://doi.org/10.17749/2313-7347/ob.gyn.rep.2023.441

![]()
101000 г. Москва, вн.тер.г. муниципальный округ Басманный, пер. Лялин, д. 11-13/1, стр. 3
Тел.: 89268786445
e-mail: digevskaya@irbis-1.ru
Обработка персональных данных






































